
The Future is in Focus: Nurturing Innovation and Collaboration in Pediatric Liver Transplantation
The WebApp is sponsored by: ![]()
Neonatal Cholestasis with Bile Duct Paucity in a Patient with a de novo EP300 Pathogenic Variant
Sulaikha Buuh1, Eva S Kahn3, Chady Meroueh4, Brendan Lanpher3, Nawras Habash2.
1Pediatrics , Mayo Clinic, Rochester, MN, United States; 2Pediatric Gastroenterology and Hepatology , Mayo Clinic, Rochester, MN, United States; 3Clinical Genomics , Mayo Clinic, Rochester, MN, United States; 4Pathology , Mayo Clinic, Rochester, MN, United States
Introduction
Neonatal cholestasis remains a diagnostic challenge. Targeted gene panels have reduced the proportion of previously unclassified cases; however, negative results do not exclude an underlying genetic etiology. Whole genome sequencing may facilitate diagnosis and identify novel genetic causes. Herein, we describe a patient with neonatal cholestasis and bile duct paucity in whom whole genome sequencing (WGS) identified a de novo, pathogenic variant in EP300, supporting an expansion of the EP300-related phenotype.
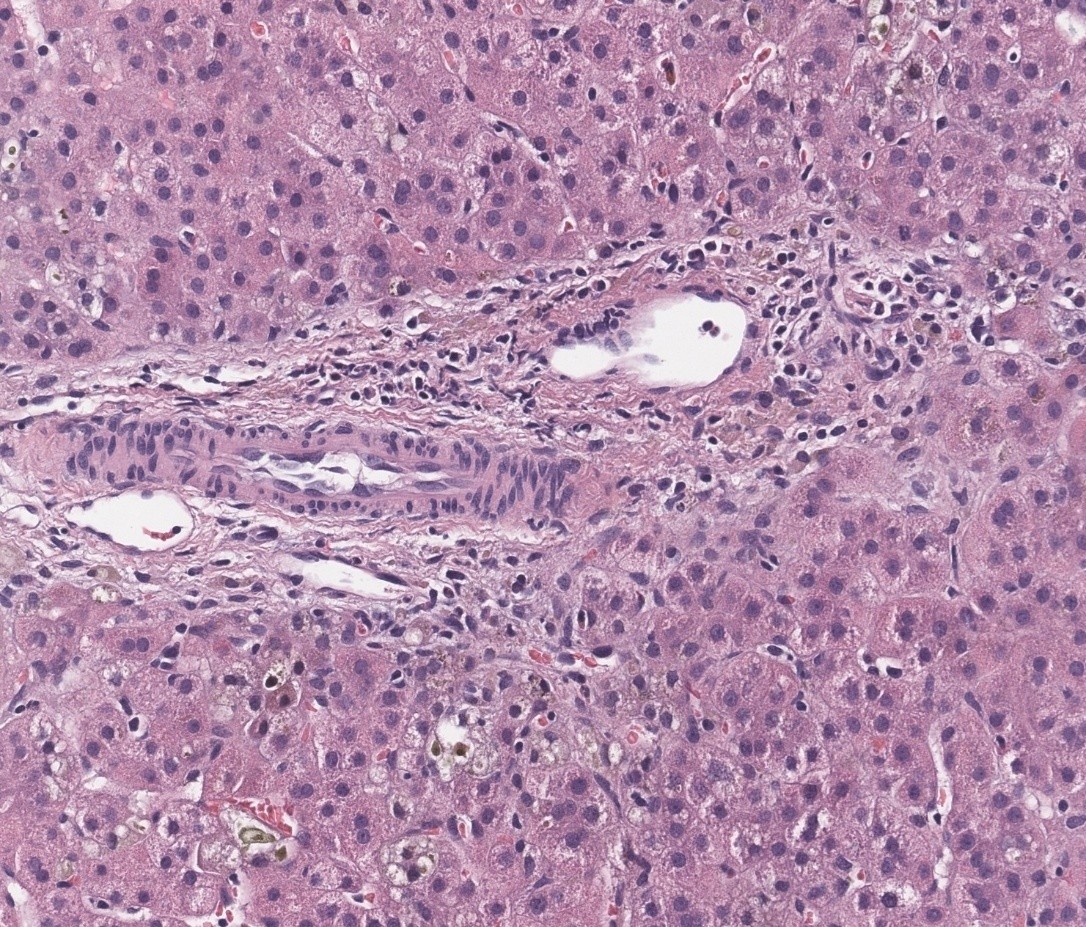
Case Description
A 17-month-old male presented at birth with high gamma glutamyl transferase cholestasis and acholic stools. Physical examination demonstrated a non-dysmorphic, jaundiced infant with mild hepatomegaly. Initial evaluation performed externally revealed a negative infectious workup, bile duct paucity on liver biopsy (Fig1), a targeted cholestasis gene panel that was unrevealing, unremarkable slit lamp examination, renal ultrasound, and spinal radiography. A non-excreting intraoperative cholangiogram prompted choledochojejunostomy, which failed to restore biliary excretion and resulted in persistent cholestasis with progressive liver disease and intractable pruritus, prompting referral to our center at 1 year of age.
The patient was referred to genetics where they underwent trio whole genome sequencing (WGS). Testing identified a de novo heterozygous, pathogenic missense variant in EP300, specifically c.4783T>G/p.Phe1595Val located in exon 30. Prior to release of results, the patient underwent liver transplantation for complications of chronic cholestasis, with marked clinical improvement.
At the time of WGS result disclosure, physical examination appreciated mild microcephaly, full cheeks, shallow orbits, arched eyebrows with prominent eyelashes, and broad thumbs and great toes. Targeted methylation analysis for the methylation signature associated with Rubinstein-Taybi syndromes 1 & 2 was abnormal with moderate confidence, while the methylation signature associated with the IDR4 domain expected with Menke-Hennekam syndromes 1& 2 was negative.
Discussion and Conclusion
Cholestasis with bile duct paucity occurs in syndromic (e.g., Alagille syndrome) as well as non-syndromic etiologies (e.g., alpha-1 antitrypsin deficiency). Pathogenic variants in EP300 are associated with EP300-related conditions including Rubinstein–Taybi syndrome
(RSTS) and Menke-Hennekam syndrome (MHS), which is phenotypically distinct from RSTS. Our described patient’s clinical phenotype and methylation data support a diagnosis of RSTS. EP300 plays a role in hepatic biogenesis and although bile duct paucity has not been previously reported in this context, the association is biologically plausible and warrants further investigation.



Email: info@splitmeeting.org
If you have any questions during the meeting, please go to the registration desk. Our emails will be monitored sporadically.
REGISTRATION DESK OPENING TIMES